Nature Communications | 清华大学生命学院张强锋课题组开发单细胞测序数据在线整合的人工智能算法
随着单细胞测序技术的发展,单细胞科学研究不断深入,规模越来越大,所研究的对象也越来越复杂。整合来源不同的单细胞测序数据,消除批次效应,进行全面挖掘和解析,是现在单细胞测序数据分析的一个基础和核心环节。目前单细胞测序数据整合面临以下几方面难题:1)不同实验样本、实验平台、建库方法乃至操作等因素带来的批次效应会在单细胞测序数据中引入非生物学噪音,干扰细胞间生物学差异的提取和解析;2)单细胞研究的规模不断扩大,百万细胞数目级别的数据对整合算法的效率提出了更高的要求;3)单细胞测序样本的类型也在不断增加,不同的单细胞测序数据集通常包括高度异质的细胞亚群;4)最后也是最新最重要的一点,如何充分重复利用大量已有数据的旧知识,对新数据进行探索和解析。目前单细胞测序数据整合算法大多基于不同批次数据间的细胞相似性来矫正批次效应,存在过度整合(尤其是整合细胞异质性差异较大的数据集)、可扩展性差、无法直接将已有模型应用到新数据集上等弊端。
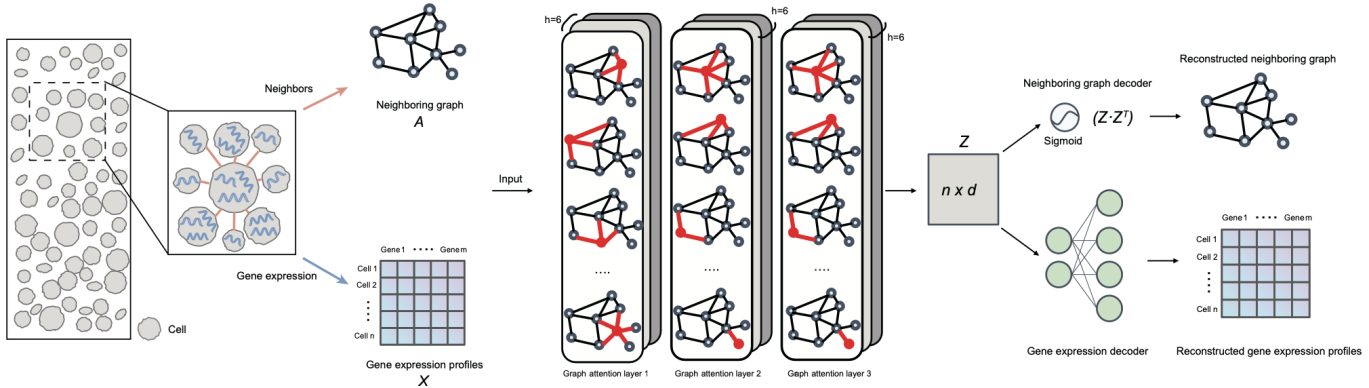
2022年10月17日,清华大学生命科学学院/结构生物学高精尖创新中心/清华-北大生命科学联合中心张强锋副教授课题组在《自然通讯》(Nature Communications)杂志在线发表题为“通过将异构数据集投影到统一的细胞嵌入空间中进行单细胞测序数据在线整合”(Online single-cell data integration through projecting heterogeneous datasets into a common cell-embedding space)的研究论文。在该研究中,他们开发了基于变分自编码器(variational autoencoder)深度学习框架的人工智能算法SCALEX,可以对单细胞测序数据进行在线整合。SCALEX采用一个批次无关的编码器和批次特异的解码器组成的非对称自编码器结构,进行大量学习得到一个高泛化性的编码器,该编码器通过将高维单细胞测序数据投射到低维细胞嵌入空间(cell embedding space),在保留生物学差异的同时消除批次效应。

图:SCALEX 模型框架
SCALEX主要有以下四点主要特征:1)相较于目前已有的单细胞测序数据整合方法,SCALEX在整合准确性上具有明显优势;2)SACLEX在百万单细胞数据量下仍保持很高的计算效率,适用于超高通量单细胞测序数据整合分析工作;3)SCALEX有效避免了单细胞测序数据整合中的过校正情形,适用于异质性高、复杂样本的整合;4)支持单细胞RNA-seq,单细胞ATAC-seq等多组学整合数据整合。这些特征使得SCALEX适用于构建单细胞图谱。开发人员整合多项研究、多个组织的单细胞数据集构建了小鼠、人以及COVID-19等三套大规模单细胞图谱。
SCALEX有一个特殊的优势,就是它的高泛化性的编码器。这个编码器可以通过单细胞测序数据投射,生成一个批次无关的统一低维细胞嵌入空间。对于新产生的数据,SCALEX不需要重新训练编码器,就可以将新数据投射到这个统一的低维细胞嵌入空间。这种整合方式被称为“在线整合”(online integration)。在线整合带来一个巨大的好处,就是很容易将新数据与原来生成的单细胞图谱等奠基性数据(需要由通过SCALEX数据整合生成)进行比较分析,从而从奠基性数据得到生物学知识方面的启发和指引,直接支持数据注释、规律验证等分析任务。另外,原有单细胞图谱的细胞内涵也在不断添加新数据的过程中,得到丰富和扩充,赋能新的生物学发现。
综上所述,该研究中,研究者们开发了SCALEX单细胞测序数据人工智能分析工具,可以将不同批次细胞的基因表达谱映射到批次无关的统一低维细胞嵌入空间中,有效消除数据中的批次效应并保留细胞间固有的生物学差异,实现不同批次数据的有效整合。SCALEX适用于图谱级别的单细胞测序数据整合,将在整个生命科学和生物医学领域正在进行的超大规模单细胞图谱等研究计划中提供基础支持。
清华大学生命科学学院张强锋副教授为本文通讯作者,清华大学生命科学学院2015级博士生熊磊(已毕业)和2018级博士生田康为该论文共同第一作者,2019级博士生李雨哲和2021级博士生宁微希对文章中的数据分析提供了重要帮助,百图生科(BioMap)研究院主任AI科学家、阿卜杜拉国王科技大学计算生物学家高欣教授参与合作研究。本工作得到国家重点研发计划、国家自然科学基金、北京市结构生物学高精尖创新中心、清华-北大生命科学联合中心、清华大学计算平台、上海期智研究院和阿卜杜拉国王科技大学研究管理办公室的支持。