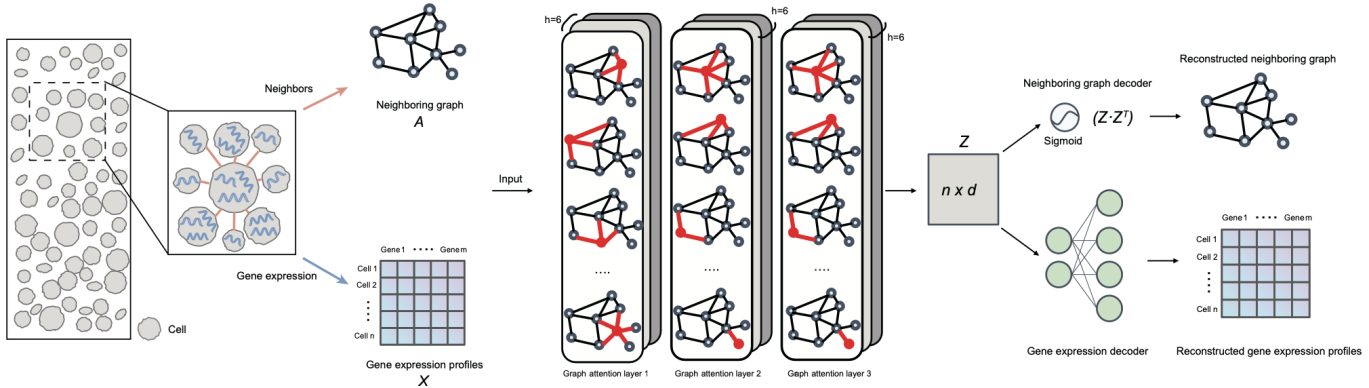
Briefings in Bioinformatics |同济大学王晨飞课题组开发单细胞CRISPR筛选数据综合分析流程SCREE
CRISPR筛选(CRISPR-screen)技术基于测序手段量化靶向目标基因的sgRNA在筛选前后的变化,目前被广泛用于筛选与特定功能表型(如细胞生长,分化,免疫耐受或耐药)相关的候选基因。虽然CRISPR筛选能在一定程度上研究基因功能,但是这一技术仍然存在很大的局限性,无法研究目标基因下游的调控机制。近年来,随着一系列高通量单细胞测序技术的出现与普及,近期的研究将CRISPR筛选与单细胞转录组测序(scRNA-seq)[1, 2]或单细胞染色质可及性(scATAC-seq)[3]等技术相结合,用于进一步探究目标基因功能及基因间的相互调控关系。这些新技术的出现为理解基因型-表型关系提供了丰富的图谱,但也带来了重大的分析挑战。
2023年3月30日,同济大学王晨飞课题组在Briefings in Bioinformatics杂志上在线发表文章SCREE: a comprehensive pipeline for single-cell multi-modal CRISPR screen data processing and analysis,开发了基于单细胞CRISPR筛选数据的综合性分析流程SCREE。

SCREE (Single-cell CRISPR screens data analysEs and perturbation modEling) 主要分为两个部分:数据预处理和下游分析。在数据预处理部分,SCREE使用双端测序的FASTQ文件作为输入,对sgRNA和mRNA/DNA均进行比对和定量。对于基于scRNA-seq的数据,SCREE可生成单细胞基因表达矩阵;对于基于scATAC-seq的数据,用户可以选择基于具有固定长度的基因组区域(bin)或是基于读长富集峰(peaks)输出单细胞矩阵。在下游分析部分,SCREE使用计数矩阵作为输入,执行一系列质量控制、可视化和分析步骤。这些步骤包括sgRNA信息可视化、单细胞质量控制、聚类和扰动富集可视化、扰动效率建模、基因调控得分估算、靶基因或增强子识别和潜在靶点的功能分析。为了方便用户对一系列分析结果进行查看,SCREE将所有结果都存放于具有良好层次结构的路径中,并且可以在HTML文件对数据的基本信息和分析结果进行可视化(图1)。
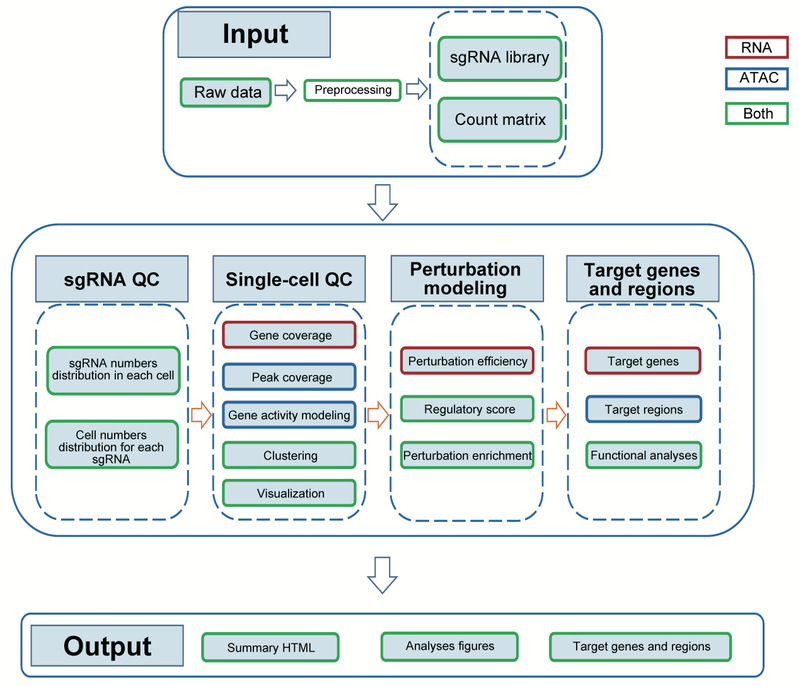
图1 SCREE工作流程示意图
为了论证SCREE在单细胞CRISPR筛选数据中应用的普适性,文章将其应用于针对基因区域进行扰动的scRNA-seq类型数据(Gene Perturbation)、过表达外源开放阅读框的scRNA-seq类型数据(Gene Overexpression)、针对增强子区域进行扰动的scRNA-seq类型数据(Enhancer Perturbation)和针对基因区域进行扰动的scATAC-seq数据(Perturb-ATAC)。在基因扰动数据中,SCREE对细胞进行聚类并在部分类群中识别出富集的扰动基因;此外,扰动效率、基因调控得分和基于基因调控得分得到的扰动间相关性三者显示出高度的一致性(图2 A-D)。在过表达开放阅读框数据中,与已有文献报道结果一致,SCREE同样能够得到LTBR基因在某一特定细胞类群中的富集,并且能够根据基因调控得分富集出相应的通路(图2 E, F)。
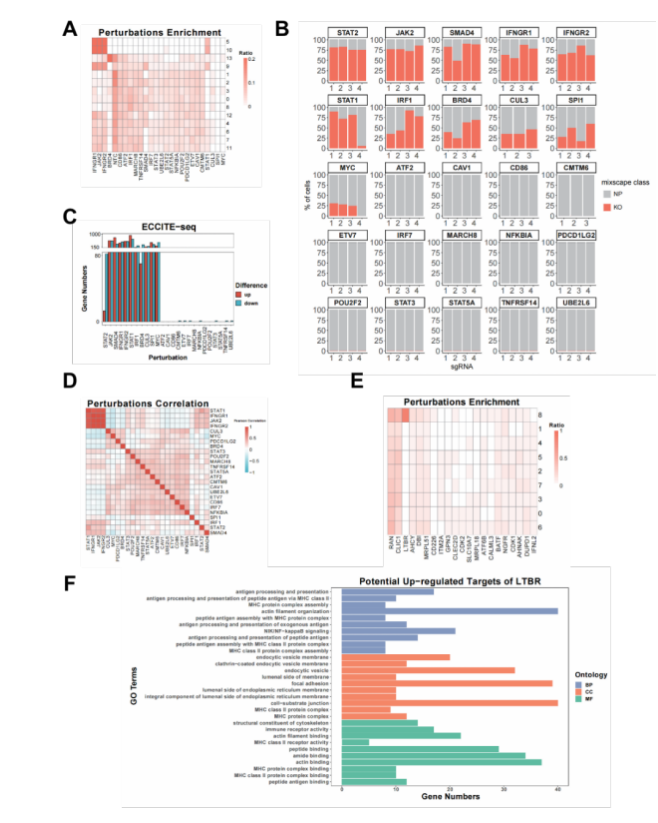
图2 SCREE在多套数据中的应用
在增强子扰动数据中,SCREE识别出每个扰动增强子周围的潜在受到其调控的基因,并进一步利用基因调控得分对增强子和这些基因间的调控关系进行了可视化,结合这些基因在扰动状态下的表达水平变化,帮助识别潜在的增强子-基因对(图3 A, B)。应用于Perturb-ATAC数据,SCREE首先将原有的基于peaks或bin的表达矩阵转变为基因活性矩阵,以执行与基因扰动数据类似的相关分析;其次,针对每个扰动基因,SCREE从原有的peaks或bin矩阵中得到与阴性对照组存在差异的区域,从这些区域中识别出潜在的增强子区域,并对该区域周围基因的基因活性和基因调控得分进行可视化,以帮助得到潜在的基因-增强子-基因调控机制(图3 C, D)。
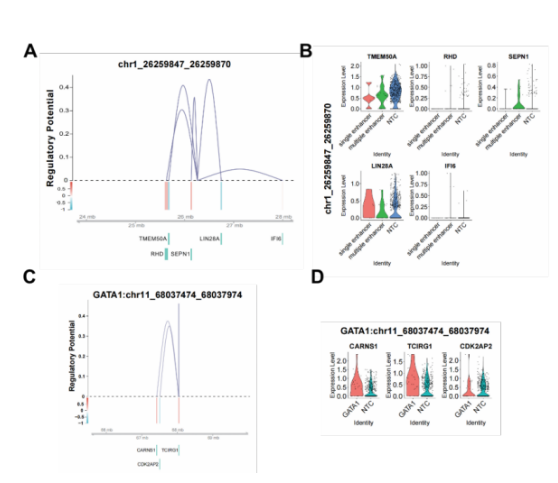
图3 SCREE在多套数据中的应用
为了验证SCREE计算基因调控得分的高效性,文章从现有公共数据中选取了包含不同细胞数目的数据集,并对其中一套数据进行了多次复制,以将细胞数目提升到百万级别。与其他基于R语言的生物信息学方法进行比较[4],SCREE具有更高的计算效率,并且能够在同等计算资源的情况下应用于细胞数据达到百万级别的数据集(图4)。
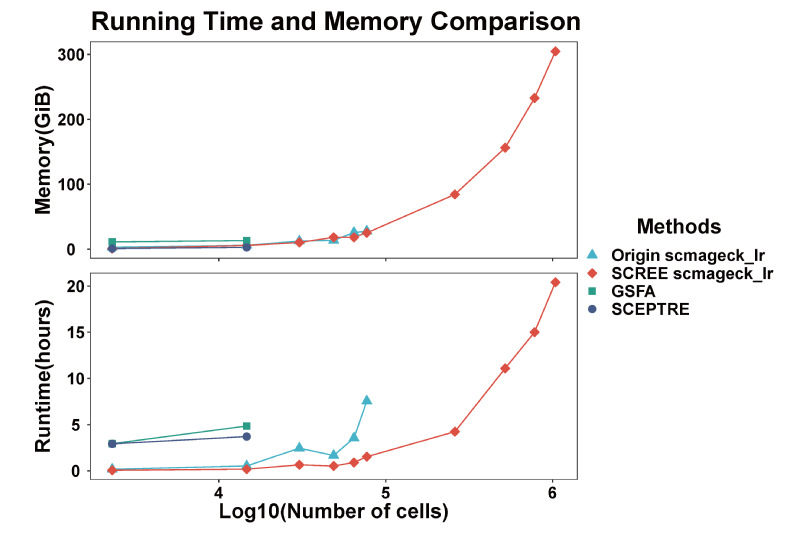
图4 SCREE基因调控得分计算效率比较
总的来说,SCREE是一种包括了序列比对和定量、数据质量控制、聚类、扰动富集可视化、扰动效率建模、基因调控得分计算、增强子识别和功能分析等功能在内的综合性分析流程,可以灵活且高效地应用于多种类型的单细胞CRISPR筛选数据。
同济大学生命科学与技术学院王晨飞教授、吴秋博士为该论文通讯作者,同济大学生命科学与技术学院博士研究生魏海霖为文章的第一作者。该项工作得到了国家自然科学基金委及上海市科委等项目的重要支持。
参考文献:
1. Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell. 2016;167(7):1853–1866.
2. Papalexi, E., Mimitou, E.P., Butler, A.W. et al. Characterizing the molecular regulation of inhibitory immune checkpoints with multimodal single-cell screens. Nat Genet 53, 322–331 (2021).
3. Pierce SE, Granja JM, Greenleaf WJ. High-throughput single-cell chromatin accessibility CRISPR screens enable unbiased identification of regulatory networks in cancer. Nat Commun. 2021 May 20;12(1):2969.
4. Yang L, Zhu Y, Yu H, Cheng X, Chen S, Chu Y, Huang H, Zhang J, Li W. scMAGeCK links genotypes with multiple phenotypes in single-cell CRISPR screens. Genome Biol. 2020 Jan 24;21(1):19.